Think of the last meal you had. Whether it be rice, pizza or shawarma, we all know better to reach our protein goal for the day. Proteins seem to be trending nowadays, with people becoming more and more health conscious. For years, we have known that proteins are good for your body. We imagine them as angels that only do good. But what happens when your benefactor turns against you? When it modifies itself to suit its needs? What if these modifications could kill you? Proteins are not all they seem, and some turn malicious and cause harm. One such example is Prions, which are deadly infectious proteins.
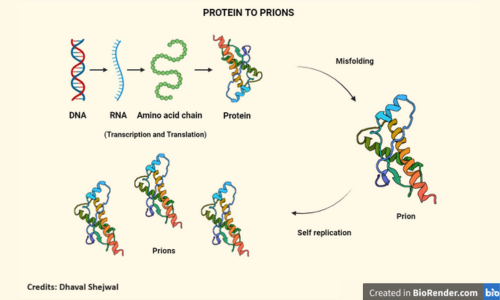
Prions are misfolded or mutated proteins, first discovered in 1982 by Stanley B. Prusiner, who won a Nobel prize for his work. These proteins cause diseases called prion diseases in humans, cattle and wide range of animal species. The surprising thing about these molecules is that they have no genetic material, neither DNA nor RNA. Yet, they can spread from one person to another, a trait that was thought impossible since DNA and RNA were believed to be the blueprints for infectious agents. Another startling fact about prions is that they can self-replicate, meaning they do not rely on any other mechanism to produce more of their kind, and without any nucleic acids (i.e. DNA and RNA), they manage to do so without any blueprint! How they do so is quite interesting. Before that, let’s understand proteins a little better. Proteins comprise molecules called amino acids, which are 20 in number. These come together in several permutations and combinations to form chains, which fold in specific ways to give rise to proteins. An ironed cotton shirt needs to be folded properly to avoid wrinkles. Similarly, the chains of amino acids need to be folded well for the protein to function. When the protein misfolds, it gives rise to prions. Prions, in turn, influence other proteins to misfold. An example could be a bad friend who slowly influences you to do wrong things. In the same way, prions influence other proteins to misfold, using themselves as a template, like a bad acquaintance whispering lies in your ears, giving themselves as examples. This leads to the formation of more Prions, and thus in this manner, they self-replicate.

Not all proteins are influenced to misfold. If that were possible, chaos would ensue within our bodies. In humans, prion diseases are caused by proteins called Prp. This protein has two forms Prpc and PrpSc. Prpc is the normal form, found in the plasma membrane of cells, especially in the central nervous system, whose exact function is still unknown. PrpSc is the prion form of the same protein and catalyzes the conversion of Prpc to PrpSc. This conversion leads to conformational changes in the Prpc structure, granting it the pathological and infectious abilities that make prions deadly.
Now, we understand what prions are, and how they become infectious. Naturally, one might wonder how do prions spread? Where could I catch this malicious protein from? Prions can be transmitted via contaminated surgical instruments, contaminated meat (tough luck, meat lovers!), and blood transfusions. Prions shed into the environment can be transmitted as well. They are also caused by genetic mutations. As mentioned before, proteins are made up of long chain of amino acids, the sequence of which is crucial. Mutations in the gene may lead to a change in the amino acid sequence which can cause the misfolding of the protein, leading to prion formation. The next generation can inherit these mutations, which is why people with a history of prion diseases in their family often get tested for the same. If you’re vegetarian, and think you are off the hook, think again since you still could get prion disease due to something called a ‘spontaneous misfolding event’. Most of the time, proteins fold in the way they are supposed to, but probability plays its funny games, and despite having all the right conditions, the protein could still misfold to give rise to a prion.

Once inside the body, the prions begin their journey by travelling to the secondary lymphoid organs, and bide their time as they replicate. Since Prpc is a protein found in our bodies, our immune system fails to recognize its mutated form as a foreign entity, thereby escaping the defence system of our body. They eventually cross over into the nervous system, and form clumps, leading to symptoms such as brain damage, dementia, hallucinations, fatigue and confusion. Prions cause human transmissible spongiform encephalopathies (also called prion diseases) which include Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker (GSS) disease, and fatal familial insomnia (FFI) among others. There are several ways to manage the disease, but unfortunately, prion diseases are almost always fatal. Once contracted, one could only wait for death since there is no cure. Things get trickier since the symptoms of prion diseases can be confused with other neurodegenerative disorders. Prion diseases are not only quite rare but also rapidly progressive, giving their victims very little time, thus their study becomes even more difficult. Yet, with the limited data they have scientists are trying to find a cure for the fatal disease. One approach is to target the chaperone molecules. It has been suspected that prions use RNA, proteoglycans and lipids to assist in their infectivity, assisting them in their crimes. Thus, by targeting these molecules, one could find a cure. Another approach comes from stopping the production of the protein itself. While the function of Prpc is unknown, its absence leaves no more Prp molecules for conversion. This can be done by targeting the production of Prpc. Molecules called antisense RNA are being developed as a cure. When a protein is formed, our DNA sends a message to the ribosomes, the protein factories, using an RNA molecule called mRNA or messenger RNA. The antisense RNA will bind to the mRNA, intercepting the message before it reaches the ribosome and thereby preventing the formation of the protein. This approach is currently in its clinical trial phase.
Prions, while deadly, have fascinated scientists for years, and have inspired more research to further our understanding of them. Their discovery changed our perspective of what an infectious agent was and what it could be. No one could fathom proteins, the components that form enzymes, the backbone of life itself could betray us. While it is unlikely that we will ever encounter prion diseases in our lives, one can marvel at nature, which created proteins to both create and destroy us.
. . .
Writer
Jwalanthi Sundaram
Editor
Tejas Nimkar
Illustrator
Dhaval Shejwal